In May I had the opportunity to give a talk at the National Cancer Institute’s Containers and Workflows Interest Group (NCI-CWIG) on “Scalable and Reproducible Genomics Data Analysis on AWS”. I’ve been giving talks like this for a while and the themes haven’t changed much over the years. I try to refresh my slides every go round, and this time through I added the following one:
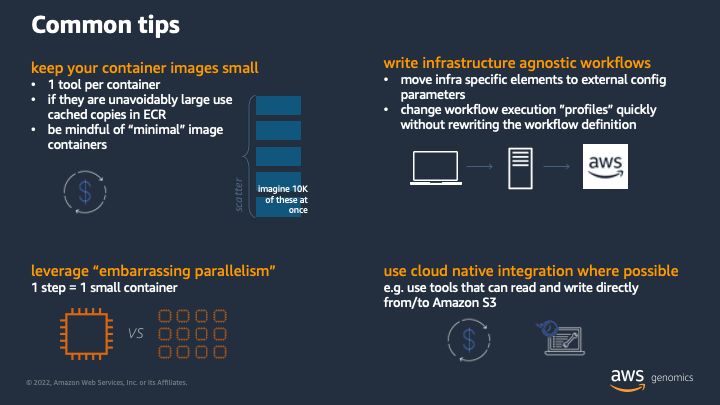
I spent about a minute or two talking through the above points during the session, but I think each warrants a bit more of a deep dive.
Keep your container images small
Containers are a means to package tools and their dependencies so that they can run anywhere. That being said, I’ve seen containers of many different sizes from miniscule (5MB) to gargantuan (>6GB).
On the large end, there are tooling distribution containers. They include many tools with the intention of being a single container to either run your entire workflow within with one compute step, or to be used for all steps of a workflow over distributed compute. I don’t recommend this for the following reasons.
ease managing tooling versions
Containers with multiple in-built tools make it difficult to manage individual tooling versions. Say you have a workflow that does the following:
tool-a (version n) → tool-b (version m) → tool-c (version p)
One day you need to update tool-b to version m+1 either to patch a critical bug or add a valuable new feature. Otherwise, you want to keep the versions of tool-a and tool-c the same. To manage this, you might try rebuilding the whole container, upgrading just tool-b. However, you might run into clashes if tool-b has shared dependencies with tool-a and tool-c. Alternatively, if you still need to run workflows that use the old version of tool-b, you now have two large container images to store and maintain.
Instead I recommend having one container image per tool. This helps to isolate concerns and reduces the amount of stuff that needs to be in each container. With each tool in their own container in the example above, updating tool-b to version m+1 is simply a matter of swapping out the old container image for tool-b with a new one, leaving tool-a and tool-c untouched.
less network bandwidth when pulling containers
Big containers will use a lot of network bandwidth to pull down. This can be problematic if (a) the compute instance a job is placed on doesn’t have much network throughput, (b) there are multiple copies of the job on the same compute instance pulling the container image, (c) the upstream image registry has limits on how much container data you can pull.
Bioinformatics workflows commonly include scatter-gather steps to leverage parallelism - e.g. processing many small intervals of a larger sequence at once using many small compute jobs. Scatters can be large - according to latest research, there are ~64,000 genes in the human genome (roughly 20,000 that code proteins) and one can imagine each being an interval to process. Now imagine the network and throttling issues with pulling a 6GB container 20,000 to 64,000 times. There are ways to partially mitigate this, like enabling container image caching on instances, using Amazon ECR pull through caching, and using instance types with more network bandwidth for compute jobs. Of course, all of these strategies are made even more effective if the job container is small to begin with.
... but avoid caveats of going too small
There are a lot of tools built with C/C++ using glibc shared libraries. The AWS CLI v2 is one of these tools. It is common for workflow engines running on AWS to bind mount the AWS CLI from the host instance into the container so that it is available for interacting with other AWS services like staging data from Amazon S3. Challenges arise when a tooling container is based on an image without glibc shared libraries as is the case with ultra-minimal base images like alpine and busybox. You can still use these ultra-minimal images, but you need to take extra steps to ensure that glibc shared libraries are available. For example, the AWS CLI v2 is distributed with the shared libraries it needs, and to make it work on an alpine based container, you can modify the LD_LIBRARY_PATH environment variable in the container environment to point to where these shared libraries are installed.
Leverage embarrassing parallelism
When running workflows in the cloud you have lots of distributed compute resources available to you. While you can use one large instance to run all steps of a workflow, this is an anti-pattern that can result in higher than expected costs.
To run an entire workflow on a single instance, you’d need to size it for the biggest step in your workflow. In the case of genomics, this is likely to be at the start of a workflow - e.g. lot’s of memory to do a reference alignment - where the rest of the workflow has relatively modest cpu and memory requirements. While the alignment step gets the resources it needs, the instance will be under utilized for the remainder of the workflow. The non-resource heavy parts of the workflow will cost more to run than they have to.
Instead, break up the workflow - run each step on its own compute instance with resources right sized for each step. There are a lot of AWS instance types to choose from, and if you connect a workflow engine to AWS Batch, AWS Batch will manage picking the right one for you based on CPU and memory (and GPU) requirements.
Write infrastructure agnostic workflows
Portability and reproducibility are key issues in scientific research. There was a whole mini-symposium at BioIT World this year devoted to FAIR principles. What this boils down to is workflows should be able to run at multiple scales and on multiple compute infrastructure - e.g. your laptop, your local HPC, your favorite cloud provider, ... and repeat for your collaborators. This matches a workflow’s deployment life cycle: initial development and small scale testing on a laptop or local cluster, production scale execution in the cloud, and sharing with others once it gets published in a scientific journal.
One should avoid hard coding infrastructure specific details into a workflow definition. Workflow languages are meant to abstract the workflow execution logic from the compute resources it runs on. For example, most languages distill a workflow task to the following:
- container image for a tool
- compute requirements like cpu, memory
- the command the container will run
- the data sources the task needs to read
- the data the task is supposed to produce
This is all fairly generic. That said, most languages also allow you to provide special per task options like:
- what job queue to run on
- how to handle retries
- special user permissions or roles to access external resources
These are infrastructural specifics and limit workflow portability if hard coded in a workflow definition. Instead, one should parameterize these as much as possible and use execution “profiles” to adapt these values to the infrastructure used at workflow runtime. For example, for a Nextflow workflow, you can do this with a nextflow.config file that looks like the following:
profiles {
stub { # use for local development and rapid iteration
process {
memory = '1 MB'
errorStrategy = 'terminate'
}
}
hpc {
process {
executor = 'sge'
queue = 'long'
clusterOptions = '-pe smp 10 -l virtual_free=64G,h_rt=30:00:00'
}
}
aws {
process {
executor = 'awsbatch'
cliPath = '/opt/aws-cli/bin/aws'
queue = 'my-spot-queue'
}
}
}
You can then invoke the workflow using one of the profiles using:
nextflow run ./myworkflow -profile aws
The profile configuration can also be easily modified to suit someone else’s environment without directly changing the workflow definition.
Use cloud native integration where possible
In addition to a wide variety of compute, running workflows on AWS also provides the ability to interact with other AWS services like Amazon RDS databases, key value stores like Amazon DynamoDB, purpose built AI/ML services like Amazon Comprehend Medical, and many more.
One common pattern to integrate with AWS from a workflow job is to call additional services using the AWS CLI. Overall, this works well, but there are a few considerations one should note when doing so. First and foremost, a workflow job needs to know where the AWS CLI installed and how to use it. You can do this by either installing the AWS CLI on the host compute and bind mounting it into the container job, or including the AWS CLI as part of the container image. That said, see my notes above on keeping container images small for associated caveats. Second, while the AWS CLI is great for scripting, for more complex operations direct integration via the AWS SDK is a better fit.
For example, a typical integration is accessing data in Amazon S3. There are lots of genomics data in Amazon S3. A common processing pattern is to stage input data from Amazon S3 onto a compute node, process it with a containerized tool, and stage the output data back to Amazon S3 ... wash and repeat for each step of a workflow. While it sounds counterintuitive, this approach is fairly economical and functions well overall. However, this approach can result in longer compute times since tools would need to wait for input data to stage in before processing can begin. This is more evident if a tool only needs to read a small portion of a larger data file (e.g. scatters over genomic intervals).
In contrast, processing can start immediately and only transfer what is necessary if tooling can read bytes of data directly from Amazon S3. Tools based on htslib can do this, so you can run something like:
samtools view -H s3://1000genomes/data/NA12878/alignment/NA12878.alt_bwamem_GRCh38DH.20150718.CEU.low_coverage.cra
Overall, this can help your workflows run faster and more efficiently (e.g. less use of node-attached storage).
Closing
There are a number of things you can do to ensure that your genomics workflows run at scale. Each of the points above are simple to implement on their own and can be combined as needed to best suit your workflow and tooling. I recommend giving each of these a try, and if you have tips of your own do share them in the comments!





Oldest comments (0)