Introduction
It has been a (long) while since I wrote my last post about the pairwise alignment of two DNA sequences using the Needleman and Wunsch-algorithm (I didn't upload that one to DEV). This was mainly because of some changes in my private life. However, I’ve now returned to bring you the promised Multiple Sequence alignment based on the previous post! It should be noted that I wrote this code almost directly after the previous post, but never got to finish it back then. I have finished it, but there is still a problem in the code I cannot fix (alloc-dealloc-mismatch). The code should finish everything as expected and export all files but expect a Segmentation fault error at the end. I am still on a basic level of C/C++ programming, so if you have any suggestions/fixes, please let me know!
The MSA uses the same principles as the pairwise alignment but will enable you to pair up multiple DNA sequences as the name suggests. There are multiple approaches to this, but my preference goes to the following: initially, we align all sequences using the pairwise alignment. This will give us an overview of which sequences are more likely to be closely related to each other and which ones are less similar to others. After determining the highest-scoring pair (which I’ll call the in-group), the alignment against the third sequence will be with the aligned sequence of the in-group. In other words: if AB is the in-group and their aligned sequence is called AB_al, then DNA sequence C will be aligned against AB_al.
All data (anonymised if applicable), used script and created visualisations can be found in my GitHub repository.
I love visualising interesting, uncommon datasets and working on projects so please let me know down in the comments if you have any request or interesting data laying around!
MSA-algorithm
As I stated in my previous post about pairwise alignments, the goal in these alignment algorithms is to get an as perfect alignment as possible. Sometimes this will result in defective alignments since Needleman-Wunsch is an outdated algorithm, but it gives you a lot of insight on how genomic alignments are done and what problems you’ll have to deal with if you want the best match! To remind you, the principles are: a perfect match on a position on two sequences gives the highest score, mismatches get penalties and gaps are usually penalised using some function that takes into account how long the gap will actually be among multiple positions.
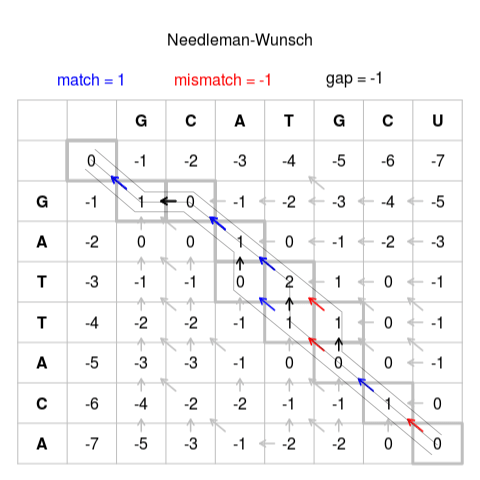
Needleman-Wunsch pairwise sequence alignment. By Slowkow - Own work, CC0
The only thing that has changed when aligning multiple sequences, is that you have to build it up iteratively from best matches to worst matches. This is why we will run pairwise alignments against all sequences and then pick the best one as our template for the other alignments. I have chosen to align only 3 sequences to keep it simple, so once you find the best pair (the in-group as I call it), you’ll only have to align the aligned in-group to the third sequence!
To boldly re-use my previous explanation about the scoring matrix: The scoring matrix shown above shows the maximal alignment score for any given sequence alignment at that point. To get the optimal alignment, you would follow the highest-scoring cells from the lower-right corner to the upper-left corner. Then you invert the given sequence to get the actual alignment. Because it aligns from the back to the front, some alignments might be somewhat unexpected as you actually read it from left to right while it is aligned from right to left.
Writing the algorithm
The top of your c++ script should contain this:
#include <iostream>
#include <fstream>
#include <string>
#include <cmath>
#include <algorithm>
#include <time.h>
using namespace std;
Some functions will need these to be executed.
Scoring methods
First things first, we’ll determine the scoring method that will be used. This will be done by using a substitution scoring matrix in which all possible combinations are given a certain score:
| C | T | A | G | |
|---|---|---|---|---|
| C | 8 | -6 | -8 | -8 |
| T | -6 | 8 | -8 | -8 |
| A | -8 | -8 | 8 | -6 |
| G | -8 | -8 | -6 | 8 |
Again, the A-G and C-T matching have a lower penalty because purine-purine (A/G) and pyrimidine-pyrimidine (C/T) mutations are biologically seen more common than other mutations. In combination with the affine gap function, the algorithm might prefer a longer gap over a gap interrupted by one of these mutations. If you compare these values to the ones in my previous post, you’ll notice some differences: I did this because I liked the results better with these values when aligning longer (~500+ nucleotides) sequences. The gap penalty will be calculated by the function gap_affinity(), which defines an “affine gap penalty”: the initial gap penalty will be higher with every directly following gap receiving a lower penalty. This ensures the algorithm to favour longer gaps over multiple singular gaps, which is also biologically seen more realistic.
It is important to understand that the previous values are not fixed values: you can set them however you want. Just make sure to check whether or not you actually get a better result out of it! If you want to do it automatically, you could build a function around the entire algorithm which changes these values iteratively or even random. Then optimize the values in such a manner that the alignment score is as high as possible. Newer algorithms based on amino acids instead of nucleotides use different matrices which are actually scientifically supported: the BLOSUM matrices are based on actual observed alignments. There are other matrices, but I’ll continue with the code!
int gap_affinity (int gap, int gap_ext, int& length){
int gap_aff = gap + (gap_ext * length);
return gap_aff;
}
Overview structure
Please note that some descriptions and functions are identical to the pairwise alignment: I included them for completeness in case you are only interested in the MSA-variant! We’ll now go through the code from the bottom to the top. This is done for a more readable structure of the code. The main() will call all main functions, gather user input and output some feedback:
int main (int argc, char** argv){
string A, B, C; // sequence to be aligned A and B
string A_al1, B_al1, C_al1, A_al2, B_al2, C_al2, A_al3, B_al3, C_al3 = ""; // aligned sequence A and B
int AB, AC, BC; // scores for initial pairwise alignments
int PW_top, score_MSA; // highest score of pairwise alignments and MSA
string MSA_ingroup, MSA_outlier, order = ""; // ingroup and outlier for MSA alignment and the order of alignments
bool print_mat; // whether to print matrices etc or just the aligned sequence
bool print_align; // if alignments should be printed
int align_nuc = 150; // amount of nucleotides per row
int a = 8; // match
int b = -6; // purine-purine / pyrimidine-pyrimidine
int c = -8; // mismatch
int gap = 4; // initial gap penalty. Gap penalty is lower than mismatch: two sequences from same species assumed.
int gap_ext = 2; // bigger gap penalties for affine gap penalty
string line; // reading in data
/*
Parameters:
Short seq:
int a = 5; // match
int b = 2; // purine-purine / pyrimidine-pyrimidine
int c = -5; // mismatch
int gap = 6; // initial gap penalty. Gap penalty is lower than mismatch: two sequences from same species assumed.
int gap_ext = 1; // bigger gap penalties for affine gap penalty
Long seq:
int a = 8; // match
int b = -6; // purine-purine / pyrimidine-pyrimidine
int c = -8; // mismatch
int gap = 4; // initial gap penalty. Gap penalty is lower than mismatch: two sequences from same species assumed.
int gap_ext = 2; // bigger gap penalties for affine gap penalty
*/
// User interface
cout << endl << "Print matrices? [0/1]" << endl;
cin >> print_mat;
cout << "Print alignments? [0/1]" << endl;
cin >> print_align;
cout << "Nucleotides per row? (150 recommended)" << endl;
cin >> align_nuc;
cout << "==============================" << endl;
// Read in the alignments
ifstream myfile ("sequences.txt");
if (myfile.is_open()){
// skip first three rows
getline (myfile,line);
getline (myfile,line);
getline (myfile,line);
getline (myfile,line);
A = line;
A.erase( A.end()-1 ); // remove whitespace at end
getline (myfile,line);
B = line;
B.erase( B.end()-1 ); // remove whitespace at end
getline (myfile,line);
C = line;
C.erase( C.end()-1 ); // remove whitespace at end
myfile.close();
}else{
cout << "Unable to open file";
return 1;
}
// Run the alignment script
int A_n = A.length();
int B_n = B.length();
int C_n = C.length();
// First pairwise alignment
cout << endl << "Initial A-B alignment:" << endl;
NW (A, B, A_al1, B_al1, A_n, B_n, a, b, c, gap, gap_ext, print_align, print_mat, align_nuc, "AB", AB);
// Second pairwise alignment
cout << endl << "Initial A-C alignment:" << endl;
NW (A, C, A_al2, C_al1, A_n, C_n, a, b, c, gap, gap_ext, print_align, print_mat, align_nuc, "AC", AC);
// Third pairwise alignment
cout << endl << "Initial B-C alignment:" << endl;
NW (B, C, B_al2, C_al2, B_n, C_n, a, b, c, gap, gap_ext, print_align, print_mat, align_nuc, "BC", BC);
cout << "Score AB: " << AB << endl;
cout << "Score AC: " << AC << endl;
cout << "Score BC: " << BC << endl;
// Print initial results
cout << endl<< "Highest PA score: ";
if (AB >= AC && AB >=BC){
PW_top = AB;
MSA_ingroup = "AB";
order = "ABC";
cout << "AB with ";
}else if(AC >= BC){
PW_top = AC;
MSA_ingroup = "AC";
order = "ACB";
cout << "AC with ";
}else{
PW_top = BC;
MSA_ingroup = "BC";
order = "BCA";
cout << "BC with ";
}
cout << PW_top << endl;
// Run second alignment
if (MSA_ingroup == "AB"){
NW_MSA (A, B, C, A_al1, B_al1, C_al1, A_n, A_n, C_n, a, b, c, gap, gap_ext, print_align, align_nuc, score_MSA, order);
}else if (MSA_ingroup == "AC"){
NW_MSA (A, C, B, A_al2, C_al2, B_al2, A_n, A_n, B_n, a, b, c, gap, gap_ext, print_align, align_nuc, score_MSA, order);
}else{
NW_MSA (B, C, A, B_al3, C_al3, A_al3, B_n, B_n, A_n, a, b, c, gap, gap_ext, print_align, align_nuc, score_MSA, order);
}
cout << "Pairwise alignment score inner group: " << PW_top << endl;
cout << "MSA score: " << score_MSA << endl;
//cout << A_n << " " << B_n << " " << C_n << endl;
// Output parameters
cout << endl << "Used parameters:" << endl;
cout << "Match = " << a << endl;
cout << "Mismatch = " << c << endl;
cout << "Purine/purine or pyrimidine/pyrimidine = " << b << endl;
cout << "Gap = -" << gap << endl;
cout << "Extended gap = -" << gap_ext << endl;
return 0;
}
As you can see, several variables are initialised and the sequences A, B and C will be read in from a file called “sequences.txt”. Just like the previously posted code, NW() is the algorithm which will align the sequences per pair. Let’s head to this function and see what it does!
First, the required matrices are created and then initialised by init(). The horizontal axis will cover sequence A and the vertical axis sequence B. After this, the alignment() function will actually align the two sequences from the back to the front: this will sometimes result in a somewhat counterintuitive global alignment. The execution time is being calculated for evaluation purposes, but can easily be skipped by editing it yourself.
After the calculations, the matrices and alignment are optionally printed and the results are exported together with the used parameters. Some print/export operations are not written in a separate function and the reason is quite simple: I sadly didn’t have the time to convert them in a function.
Printing of the alignment is done using a more biologically relevant method: nucleotides n through n+align_nuc of aligned sequences A and B are printed below each other iteratively, giving more insight in the alignment.
Note that the prefix determines which alignment we are executing: A-B, A-C or B-C.
// Initiate matrices, align and export
int** NW (string A, string B, string& A_al, string& B_al, int A_n, int B_n, int a, int b, int c, int gap, int gap_ext, bool print_align, bool print_mat, int align_nuc, string prefix, int& paired_score){
// Create alignment matrix
int** M = new int* [B_n+1];
for( int i = 0; i <= B_n; i++ ){
M[i] = new int [A_n+1];
}
// Create traceback matrix
char** M_tb = new char* [B_n+1];
for( int i = 0; i <= B_n; i++ ){
M_tb[i] = new char [A_n+1];
}
//clock_t t; // for timing execution
//t = clock(); // get time of start
// Initialize traceback and F matrix (fill in first row and column)
init (M, M_tb, A_n, B_n, gap, gap_ext);
// Create alignment
alignment (M, M_tb, A, B, A_al, B_al, A_n, B_n, a, b, c, gap, gap_ext);
//t = clock() - t; // get time when finished
int score = M[B_n][A_n]; // get alignment score
if(print_mat == 1){
print_mtx(M, A, B, A_n, B_n);
print_tb(M_tb, A, B, A_n, B_n);
}
if(print_align == 1){
cout << endl << "Alignments:" << endl;
int start = 0; // start of new line for printing alignments
int cntr = 0; // iterator for printing alignments
int Al_n = A_al.length(); // length of alignment
do{
cout << start+1 << " A: ";
for (cntr = start; cntr < start+align_nuc; cntr++){
if(cntr < Al_n){
cout << A_al[cntr];
}else{
break;
}
}
cout << " " << cntr << endl << start+1 << " B: ";
for (cntr = start; cntr < start+align_nuc; cntr++){
if(cntr < Al_n){
cout << B_al[cntr];
}else{
break;
}
}
cout << " " << cntr << endl << endl;
start += align_nuc;
}while(start <= Al_n);
}
cout << "Alignment score: " << score << endl;
paired_score = score;
// Show runtime
//cout << "Alignment took " << t << "clicks (" << (t)/CLOCKS_PER_SEC << "seconds).\n";
// Export alignment to file
cout << "Exporting results..." << endl;
ofstream myfile;
if (prefix == "AB"){
myfile.open ("AB_alignment.txt");
}else if (prefix == "AC"){
myfile.open ("AC_alignment.txt");
}else if (prefix == "BC"){
myfile.open ("BC_alignment.txt");
}
myfile << "Used parameters:\n";
myfile << "Match = " << a << "\n";
myfile << "Mismatch = " << c << "\n";
myfile << "Purine/purine or pyrimidine/pyrimidine = " << b << "\n";
myfile << "Gap = -" << gap << "\n";
myfile << "Extended gap = -" << gap_ext << "\n";
//myfile << "Average elapsed time = " << t << " - " << (t)/CLOCKS_PER_SEC << " [clicks - seconds]." << endl << endl;
myfile << "Input A:\n" << A << "\n";
myfile << "Input B:\n" << B << "\n";
myfile << "\nAlignment:\n";
int start = 0; // start of new line for printing alignments
int cntr = 0; // iterator for printing alignments
int Al_n = A_al.length(); // length of alignment
do{
myfile << start+1 << " A: ";
for (cntr = start; cntr < start+align_nuc; cntr++){
if(cntr < Al_n){
myfile << A_al[cntr];
}else{
break;
}
}
myfile << " " << cntr << "\n" << start+1 << " B: ";
for (cntr = start; cntr < start+align_nuc; cntr++){
if(cntr < Al_n){
myfile << B_al[cntr];
}else{
break;
}
}
myfile << " " << cntr << "\n\n";
start += align_nuc;
}while(start <= Al_n);
myfile << "Alignment score: " << score << "\n";
myfile.close();
// Export scoring matrix to file
if (prefix == "AB"){
myfile.open ("AB_scoring.txt");
}else if (prefix == "AC"){
myfile.open ("AC_scoring.txt");
}else if (prefix == "BC"){
myfile.open ("BC_scoring.txt");
}
myfile << "-\t-\t";
for( int j = 0; j < A_n; j++ ){
myfile << A[j] << "\t";
}
myfile << "\n-\t";
for( int i = 0; i <= B_n; i++ ){
int j = 0;
if( i > 0){
myfile << B[i-1] << "\t";
}
do{
myfile << M[i][j] << "\t";
j++;
}while(j < A_n);
myfile << M[i][j] << "\n";
}
myfile.close();
// Export traceback matrix to file
if (prefix == "AB"){
myfile.open ("AB_traceback.txt");
}else if (prefix == "AC"){
myfile.open ("AC_traceback.txt");
}else if (prefix == "BC"){
myfile.open ("BC_traceback.txt");
}
myfile << "-\t-\t";
for( int j = 0; j < A_n; j++ ){
myfile << A[j] << "\t";
}
myfile << "\n-\t";
for( int i = 0; i <= B_n; i++ ){
int j = 0;
if( i > 0){
myfile << B[i-1] << "\t";
}
do{
myfile << M_tb[i][j] << "\t";
j++;
}while(j < A_n);
myfile << M_tb[i][j] << "\n";
}
myfile.close();
cout << "Results exported!" << endl;
// Free memory
for( int i = 0; i <= B_n; i++ ) delete M[i];
delete[] M;
for( int i = 0; i <= B_n; i++ ) delete M_tb[i];
delete[] M_tb;
return 0;
}
The init() function already creates the first column and row, filling them with a hardcoded affine gap function. The traceback matrix automatically gets a horizontal or vertical line, indicating a step left or up (both gaps, but in either B or A respectively). The rest of the matrix will be filled by the alignment() function, which is discussed next.
// Initialise scoring matrix with first row and column
void init (int **M, char** M_tb, int A_n, int B_n, int gap, int gap_ext){
M[0][0] = 0;
M_tb[0][0] = 'n';
int i=0, j=0;
for( j = 1; j <= A_n; j++ ){
M[0][j] = - ( gap + ( gap_ext * (j - 1) ) ); // manually apply affine gap
M_tb[0][j] = '-';
}
for( i = 1; i <= B_n; i++ ){
M[i][0] = - ( gap + ( gap_ext * (i - 1) ) ); // manually apply affine gap
M_tb[i][0] = '|';
}
}
Aligning the sequences
The alignment is made by the function alignment(), which also takes the gap penalty as variable to feed into the affine gap function. First, the algorithm scores all possible alignment possibilities in the scoring matrix using the substitution scoring matrix. Secondly, the traceback matrix is created, based on the scoring matrix: From the lower-right corner, the highest-scoring cell directly above, diagonally left or left indicates the next step in the alignment up to the upper left corner. This will lead to the final, most optimal global alignment. Lastly, the traceback matrix is used to create the backward alignments: it follows the traceback along with the highest-scoring cells from the lower-right corner to the upper-left corner, introducing gaps in sequence A or B when not going diagonal.
// Needleman and Wunsch algorithm
int alignment (int **M, char** M_tb, string A, string B, string& A_al, string& B_al, int A_n, int B_n, int a, int b, int c, int gap, int gap_ext){
int x = 0, y = 0;
int scU, scD, scL; // scores for respectively cell above, diagonal and left
char ptr, nuc;
int i = 0, j = 0;
int length = 0; // initial gap length
// create substitution scoring matrix
const int s[4][4] = {
{ a, b, c, c },
{ b, a, c, c },
{ c, c, a, b },
{ c, c, b, a }
};
for( i = 1; i <= B_n; i++ ){
for( j = 1; j <= A_n; j++ ){
nuc = A[j-1];
switch( nuc )
{
case 'C': x = 0; break;
case 'T': x = 1; break;
case 'A': x = 2; break;
case 'G': x = 3;
}
nuc = B[i-1];
switch( nuc )
{
case 'C': y = 0; break;
case 'T': y = 1; break;
case 'A': y = 2; break;
case 'G': y = 3;
}
scU = M[i-1][j] - gap_affinity(gap, gap_ext, length); // get score if trace would go up
scD = M[i-1][j-1] + s[x][y]; // get score if trace would go diagonal
scL = M[i][j-1] - gap_affinity(gap, gap_ext, length); // get score if trace would go left
M[i][j] = max_score (scU, scD, scL, &ptr, length); // get max score for current optimal global alignment
M_tb[i][j] = ptr;
}
}
i--; j--;
while( i > 0 || j > 0 ){
switch( M_tb[i][j] )
{
case '|' : A_al += '-';
B_al += B[i-1];
i--;
break;
case '\\': A_al += A[j-1];
B_al += B[i-1];
i--; j--;
break;
case '-' : A_al += A[j-1];
B_al += '-';
j--;
}
}
reverse( A_al.begin(), A_al.end() );
reverse( B_al.begin(), B_al.end() );
return 0 ;
}
For the printing of the matrices, two functions were used from the original, referenced code:
// Print the scoring matrix
void print_mtx (int** M, string A, string B, int A_n, int B_n){
cout << " ";
for( int j = 0; j < A_n; j++ ){
cout << A[j] << " ";
}
cout << "\n ";
for( int i = 0; i <= B_n; i++ ){
if( i > 0 ){
cout << B[i-1] << " ";
}
for( int j = 0; j <= A_n; j++ ){
cout.width( 3 );
cout << M[i][j] << " ";
}
cout << endl;
}
cout << endl;
}
// Print the traceback matrix
void print_tb (char** M_tb, string A, string B, int A_n, int B_n){
cout << " ";
for( int j = 0; j < A_n; j++ ){
cout << A[j] << " ";
}
cout << "\n ";
for( int i = 0; i <= B_n; i++ ){
if( i > 0 ){
cout << B[i-1] << " ";
}
for( int j = 0; j <= A_n; j++ ){
cout.width( 3 );
cout << M_tb[i][j] << " ";
}
cout << endl;
}
cout << endl;
}
Multiple Sequence alignment
Now for the MSA-part. The highest-scoring pairwise alignment will be called the “in-group”. It is checked in lines 736 through 742 in the main():
// Print initial results
cout << endl<< "Highest PA score: ";
if (AB >= AC && AB >=BC){
PW_top = AB;
MSA_ingroup = "AB";
order = "ABC";
cout << "AB with ";
}else if(AC >= BC){
PW_top = AC;
MSA_ingroup = "AC";
order = "ACB";
cout << "AC with ";
}else{
PW_top = BC;
MSA_ingroup = "BC";
order = "BCA";
cout << "BC with ";
}
cout << PW_top << endl;
The second alignment with the in-group to the third sequence is done by NW_MSA(). It will align the sequences using alignment_MSA() to actually align the sequences. C will be aligned to the aligned sequences A and B. To compensate for the fact that we check against two sequences, the score is divided by 2 (simply getting the mean). After alignment, the output will be three rows under each other showing the alignment as usual.
// Needleman and Wunsch algorithm adjusted for MSA
int alignment_MSA (int **M, char** M_tb, string A, string B, string C, string& A_al, string& B_al, string& C_al, int A_n, int B_n, int C_n, int a, int b, int c, int gap, int gap_ext){
int x1 = 0, x2 = 0, y = 0;
int scU, scD, scL; // scores for respectively cell above, diagonal and left
char ptr, nuc;
int i = 0, j = 0;
int length = 0; // initial gap length
bool inner_gap; // if gap in inner sequence is present
// A = sequence 1 of 2 from ingroup
// B = sequence 2 of 2 from ingroup
// C = sequence outgroup
// create substitution scoring matrix
const int s[4][4] = {
{ a, b, c, c },
{ b, a, c, c },
{ c, c, a, b },
{ c, c, b, a }
};
for( i = 1; i <= C_n; i++ ){
for( j = 1; j <= A_n; j++ ){
// Initialise some values
inner_gap = 0;
scD = 0;
// Alignment outer sequence with inner sequence 1
nuc = A[j-1];
switch( nuc )
{
case 'C': x1 = 0; break;
case 'T': x1 = 1; break;
case 'A': x1 = 2; break;
case 'G': x1 = 3; break;
case '-': inner_gap += 1; break; // SM not needed when gap
}
// Alignment outer sequence with inner sequence 2
nuc = B[j-1];
switch( nuc )
{
case 'C': x2 = 0; break;
case 'T': x2 = 1; break;
case 'A': x2 = 2; break;
case 'G': x2 = 3; break;
case '-': inner_gap += 1; break; // SM not needed when gap
}
nuc = C[i-1];
switch( nuc )
{
case 'C': y = 0; break;
case 'T': y = 1; break;
case 'A': y = 2; break;
case 'G': y = 3;
}
scD = M[i-1][j-1] + (s[x1][y] + s[x2][y])/2; // get score if trace would go diagonal (inner sequences with outer sequence), compensated for double match score
scU = M[i-1][j] - gap_affinity(gap, gap_ext, length); // get score if trace would go up
scL = M[i][j-1] - gap_affinity(gap, gap_ext, length); // get score if trace would go left
scU += 0.5*(inner_gap * gap); // compensate the gap penalty with a bonus if the inner sequences also contain a gap at this location
M[i][j] = max_score (scU, scD, scL, &ptr, length); // get max score for current optimal global alignment
M_tb[i][j] = ptr;
}
}
i--; j--;
while( i > 0 || j > 0 ){
switch( M_tb[i][j] )
{
case '|' : A_al += '-';
B_al += '-';
C_al += C[i-1];
i--;
break;
case '\\': A_al += A[j-1];
B_al += B[j-1];
C_al += C[i-1];
i--; j--;
break;
case '-' : A_al += A[j-1];
B_al += B[j-1];
C_al += '-';
j--;
}
k++ ;
}
reverse( A_al.begin(), A_al.end() );
reverse( B_al.begin(), B_al.end() );
reverse( C_al.begin(), C_al.end() );
return 0 ;
}
// Initiate matrices, align and export
int** NW_MSA (string A, string B, string C, string& A_al, string& B_al, string& C_al, int A_n, int B_n, int C_n, int a, int b, int c, int gap, int gap_ext, bool print_align, int align_nuc, int& msa_score, string order){
// A = sequence 1 of 2 from ingroup
// B = sequence 2 of 2 from ingroup
// C = sequence outgroup
// Create alignment matrix
int** M = new int* [C_n+1];
for( int i = 0; i <= C_n; i++ ){
M[i] = new int [A_n+1];
}
// Create traceback matrix
char** M_tb = new char* [C_n+1];
for( int i = 0; i <= C_n; i++ ){
M_tb[i] = new char [A_n+1];
}
//clock_t t; // for timing execution
//t = clock(); // get time of start
// Initialize traceback and F matrix (fill in first row and column)
init (M, M_tb, A_n, C_n, gap, gap_ext);
// Create alignment
alignment_MSA (M, M_tb, A, B, C, A_al, B_al, C_al, A_n, B_n, C_n, a, b, c, gap, gap_ext);
//t = clock() - t; // get time when finished
int score = M[C_n][A_n]; // get alignment score
if(print_align == 1){
cout << endl << "Order of inputs: " << order << endl;
cout << "Alignments:" << endl;
int start = 0; // start of new line for printing alignments
int cntr = 0; // iterator for printing alignments
int Al_n = A_al.length(); // length of alignment
do{
cout << start+1 << " A: ";
for (cntr = start; cntr < start+align_nuc; cntr++){
if(cntr < Al_n){
cout << A_al[cntr];
}else{
break;
}
}
cout << " " << cntr << endl << start+1 << " B: ";
for (cntr = start; cntr < start+align_nuc; cntr++){
if(cntr < Al_n){
cout << B_al[cntr];
}else{
break;
}
}
cout << " " << cntr << endl << start+1 << " C: ";
for (cntr = start; cntr < start+align_nuc; cntr++){
if(cntr < Al_n){
cout << C_al[cntr];
}else{
break;
}
}
cout << " " << cntr << endl << endl;
start += align_nuc;
}while(start <= Al_n);
}
cout << "Alignment score: " << score << endl;
msa_score = score;
// Show runtime
//cout << "Alignment took " << t << "clicks (" << (t)/CLOCKS_PER_SEC << "seconds).\n";
// Export alignment to file
cout << "Exporting results..." << endl;
ofstream myfile;
myfile.open ("MSA_alignment.txt");
myfile << "Used parameters:\n";
myfile << "Match = 3\n";
myfile << "Mismatch = -3\n";
myfile << "Purine/purine or pyrimidine/pyrimidine = 1\n";
myfile << "Gap = -" << gap << "\n";
myfile << "Extended gap = -" << gap_ext << "\n";
//myfile << "Average elapsed time = " << t << " - " << (t)/CLOCKS_PER_SEC << " [clicks - seconds]." << endl << endl;
myfile << "Input A:\n" << A << "\n";
myfile << "Input B:\n" << B << "\n";
myfile << "Input C:\n" << C << "\n";
myfile << "Order of inputs: " << order << "\n";
myfile << "\nAlignment:\n";
int start = 0; // start of new line for printing alignments
int cntr = 0; // iterator for printing alignments
int Al_n = A_al.length(); // length of alignment
do{
myfile << start+1 << " A: ";
for (cntr = start; cntr < start+align_nuc; cntr++){
if(cntr < Al_n){
myfile << A_al[cntr];
}else{
break;
}
}
myfile << " " << cntr << "\n" << start+1 << " B: ";
for (cntr = start; cntr < start+align_nuc; cntr++){
if(cntr < Al_n){
myfile << B_al[cntr];
}else{
break;
}
}
myfile << " " << cntr << "\n" << start+1 << " C: ";
for (cntr = start; cntr < start+align_nuc; cntr++){
if(cntr < Al_n){
myfile << C_al[cntr];
}else{
break;
}
}
myfile << " " << cntr << "\n\n";
start += align_nuc;
}while(start <= Al_n);
myfile << "Alignment score: " << score << "\n";
myfile.close();
// Export scoring matrix to file
myfile.open ("MSA_scoring.txt");
myfile << "-\t-\t";
for( int j = 0; j < A_n; j++ ){
myfile << A[j] << "\t";
}
myfile << "\n-\t-\t";
for( int j = 0; j < B_n; j++ ){
myfile << B[j] << "\t";
}
myfile << "\n-\t";
for( int i = 0; i <= C_n; i++ ){
int j = 0;
if( i > 0){
myfile << C[i-1] << "\t";
}
do{
myfile << M[i][j] << "\t";
j++;
}while(j < A_n);
myfile << M[i][j] << "\n";
}
myfile.close();
// Export traceback matrix to file
myfile.open ("MSA_traceback.txt");
myfile << "-\t-\t";
for( int j = 0; j < A_n; j++ ){
myfile << A[j] << "\t";
}
myfile << "\n-\t-\t";
for( int j = 0; j < B_n; j++ ){
myfile << B[j] << "\t";
}
myfile << "\n-\t";
for( int i = 0; i <= C_n; i++ ){
int j = 0;
if( i > 0){
myfile << C[i-1] << "\t";
}
do{
myfile << M_tb[i][j] << "\t";
j++;
}while(j < A_n);
myfile << M_tb[i][j] << "\n";
}
myfile.close();
cout << "Results exported!" << endl;
for( int i = 0; i <= B_n; i++ ) delete M[i];
delete[] M;
for( int i = 0; i <= B_n; i++ ) delete M_tb[i];
delete[] M_tb;
return 0;
}
This concludes all the necessary code for this script. Just like with the pairwise alignment script, it is up to you whether or not you want to export data and show messages: simply disable them if desired. For longer sequences, disable the printing of the matrices to prevent a big clutter on your screen.
Data example
In the simpler pairwise alignment, I didn’t talk about the sequence.txt file. But now I will!
To start off, the first three lines hold the lengths of sequence 1, 2 and 3 respectively. The sequences themselves will be on lines 4 through 6. These were randomly generated, so remember that there’s no real solution to this specific alignment.
This file is also included in my repository.
For the complete script, please see my GitHub repository.





Oldest comments (0)